Study Sheet on Calibration
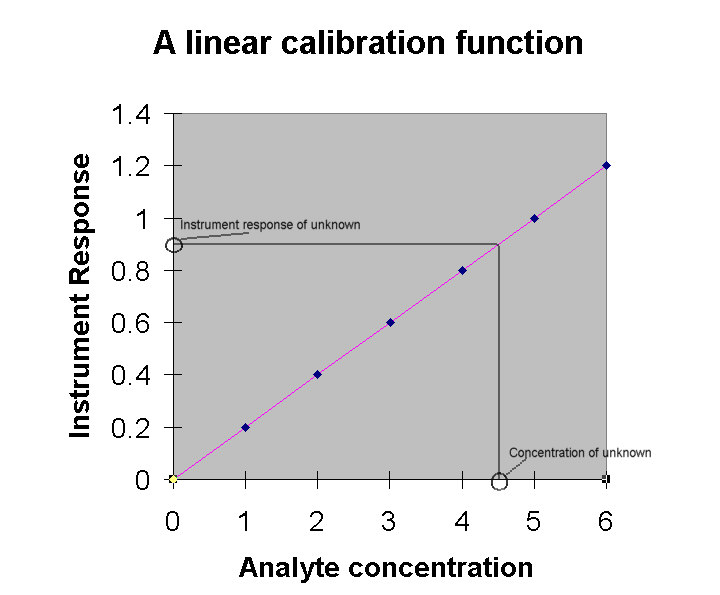 Calibration is the process
of assigning a value, usually in concentration units, to an instrument
response. For example, you might "calibrate" the response
of a UV-visible absorption spectrometer (which is in units of
absorbed light) by placing different "known" concentrations
of analyte in a cell in the light path and establishing the instrument
response per unit concentration. This "response function"
should ideally be a straight line that passes directly through
the origin. Why? Because it is unlikely that the unknown concentration,
which you will determine by extrapolating the observed "unknown
absorbance" over to the calibration function on the graph,
will fall exactly on one of the known calibration points. Therefore
you must know the calibration function exactly. A function of
a line is exactly known, and physical phenomena that produce a
linear response can be exactly modeled. On the other hand, if
the calibration function was nonlinear, it would not be as easy
to define and more sensitive to changes in physical phenomena.
There would be greater error on any concentration determined from
a nonlinear instrument response. So, what you want and what you
most often see is a linear calibration function in analytical
chemistry.
Calibration is the process
of assigning a value, usually in concentration units, to an instrument
response. For example, you might "calibrate" the response
of a UV-visible absorption spectrometer (which is in units of
absorbed light) by placing different "known" concentrations
of analyte in a cell in the light path and establishing the instrument
response per unit concentration. This "response function"
should ideally be a straight line that passes directly through
the origin. Why? Because it is unlikely that the unknown concentration,
which you will determine by extrapolating the observed "unknown
absorbance" over to the calibration function on the graph,
will fall exactly on one of the known calibration points. Therefore
you must know the calibration function exactly. A function of
a line is exactly known, and physical phenomena that produce a
linear response can be exactly modeled. On the other hand, if
the calibration function was nonlinear, it would not be as easy
to define and more sensitive to changes in physical phenomena.
There would be greater error on any concentration determined from
a nonlinear instrument response. So, what you want and what you
most often see is a linear calibration function in analytical
chemistry.
A line can be described by an equation: y = mx + b, where m
is the slope of the line in terms of Dy/Dx (i.e., change in y/change in x). The
intercept with the line of the y-axis is b. In terms of the graph
shown above, we can convert y = mx + b to the terms:
instrument response = (slope of the calibration function)
x (analyte concentration) + blank concentration.
The intercept is the "blank concentration" since
it is the instrument response corresponding to an analyte concentration
of zero. In the graph shown above, the calibration function is
established based on 7 points as shown here:
|
Concentration |
Instrument Response |
|
0 |
0.000 |
|
1 |
0.200 |
|
2 |
0.400 |
|
3 |
0.600 |
|
4 |
0.800 |
|
5 |
1.000 |
|
6 |
1.200 |
The slope in this example would be 0.200 and the intercept
would be 0. The equation of the line would therefore be:
y = 0.200x
Plug in a few values of x (instrument response) to the formula
and you will see that it applies to the data set in the table
on the previous page.
Normally, the points from which the calibration function is
determined do not fit so well as shown in the previous table.
In such a case, you use the linear regression function of a calculator
or spreadsheet to give you a line of best fit. This calibration
line will be defined by a slope and intercept, which will be given
to you in the equation of the line. Let’s plot the following
data set using the regression function of Excel (data analysis,
regression):
|
Concentration |
Instrument Response |
|
0 |
0.025 |
|
1 |
0.217 |
|
2 |
0.388 |
|
3 |
0.634 |
|
4 |
0.777 |
|
5 |
1.011 |
|
6 |
1.166 |
 You will note from the graph on the right that there
is a considerable amount of scatter around the regression line,
and that the intercept of the line on line on the y (instrument
response) axis is not zero. This is the normal situation for analytical
data and the result is that the use of a calibration line causes
a certain amount of uncertainty in the concentrations determined
from the calibration curve. This is discussed more fully in your
textbook.
You will note from the graph on the right that there
is a considerable amount of scatter around the regression line,
and that the intercept of the line on line on the y (instrument
response) axis is not zero. This is the normal situation for analytical
data and the result is that the use of a calibration line causes
a certain amount of uncertainty in the concentrations determined
from the calibration curve. This is discussed more fully in your
textbook.
If, for example, we found that an unknown solution gave an
instrument response of 0.254, we would enter that as y in the
equation and solve for x:
0.254 = 0.1929x + 0.024
x = 1.15
What can go wrong with this "direct calibration"
method? Two things: additive errors and multiplicative errors.
An additive error is one that changes the intercept of the calibration
function. Perhaps in the process of measuring the absorbance of
a series of standards using a UV-vis absorption spectrophotometer,
you unknowingly place a fingerprint on the absorption cell. Instead
of a plot as shown above, you get a line that is offset from the
origin on the y-axis by the instrument response resulting from
the fingerprint. It will be the same at all analyte concentrations.
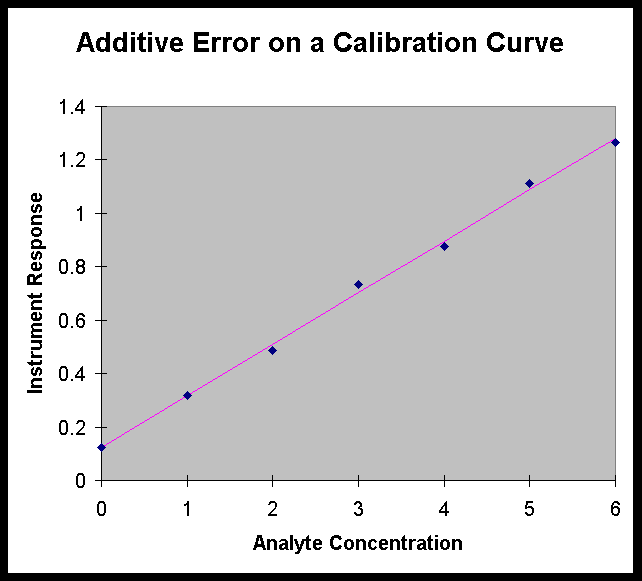 It is difficult to correct for an additive error
if you don’t know that it exists. For example, in the calibration
curve shown at the right, if the standards were measured in the
cuvette with the fingerprint, but the cuvette was cleaned before
the samples were measured, all the sample measurements would be
in error by the instrument response due to the fingerprint. To
the analyst, the calibration curve would look like the graph on
the previous page (since the instrument zero is set with the standard
blank), while it "really" looks like the graph on the
right.
It is difficult to correct for an additive error
if you don’t know that it exists. For example, in the calibration
curve shown at the right, if the standards were measured in the
cuvette with the fingerprint, but the cuvette was cleaned before
the samples were measured, all the sample measurements would be
in error by the instrument response due to the fingerprint. To
the analyst, the calibration curve would look like the graph on
the previous page (since the instrument zero is set with the standard
blank), while it "really" looks like the graph on the
right.
Careful experimental observations can sometimes detect additive
errors. For example, in the aforementioned situation, if a sample
blank were measured along with the samples, it would produce a
negative instrument response, since there would be no fingerprint
on the cuvette. That would be an immediate clue that there was
an additive error in the standard measurements.
Another way to detect such an error is to repeat the calibration
after all samples have been measured. Repetition is the key to
good analytical results and the fact that the calibration line
has shifted will become obvious when the standard measurements
are repeated with a clean cell.
Additive errors can also result from the presence of a molecule
(other than the analyte) in the sample that produces an instrument
response (i.e., absorbs light) at the same wavelength as the molecule
you are trying to determine. This extra absorption by the interfering
molecule will make it appear that there is more of the molecule
you are trying to measure than there actually is. This is a far
more difficult problem to solve, although there are instrumental
methods to deal with it.
Multiplicative interferences are those which change the slope
of the calibration line. One example of a multiplicative interference
might be if the cuvette used for the standards had a longer path
length that that used for the samples. You may recall that in
absorption spectrophotometry:
Absorbance = (molar absorbtivity) x (cell path length)
x (concentration)
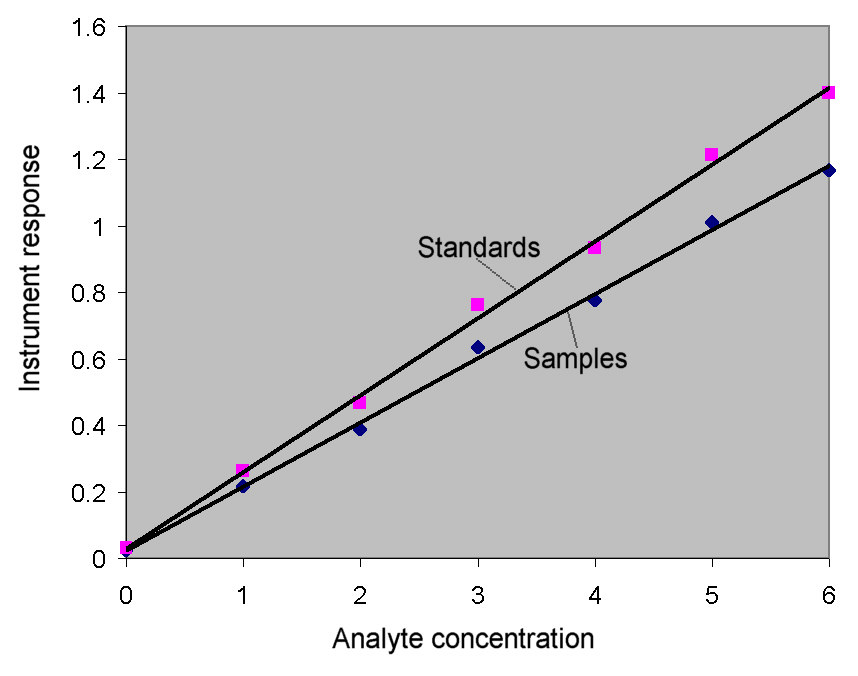 Therefore, since the cuvette used for the standards
has a longer path length, the sample absorbances will appear to
represent concentrations that are lower than they actually are.
The error on each sample measurement would constant on a relative
basis (i.e., a 10% relative error … 10% too low.) This can
be seen from the graph at the right. The curve with the greater
slope is prepared using the standards (with the longer path length).
Measurements of sample absorbance compared to this curve will
give a concentration that is too low. The curve to which the samples
should be compared has a lower slope. This curve would be generated
from standards measured in the same cuvette as was used for the
samples. The relative error between the two curves is approximately
10%.
Therefore, since the cuvette used for the standards
has a longer path length, the sample absorbances will appear to
represent concentrations that are lower than they actually are.
The error on each sample measurement would constant on a relative
basis (i.e., a 10% relative error … 10% too low.) This can
be seen from the graph at the right. The curve with the greater
slope is prepared using the standards (with the longer path length).
Measurements of sample absorbance compared to this curve will
give a concentration that is too low. The curve to which the samples
should be compared has a lower slope. This curve would be generated
from standards measured in the same cuvette as was used for the
samples. The relative error between the two curves is approximately
10%.
The change in slope of the analytical curve caused by systematic
errors such as using different cuvette path lengths can be referred
to as a change in sensitivity … that is, a change in the
instrument response per unit concentration. Such effects can be
caused by other components of a sample matrix that enhance or
depress the instrument response of the analyte in the sample matrix
as compared to the instrument response of the analyte in the standards.
How do you correct for multiplicative errors? The best way
is to use the method of standard addition, in which you add a
known concentration of analyte to the unknown sample and then
compare the increase of instrument response caused by the addition
of the analyte to the instrument response observed for that concentration
of analyte in the calibration standards.
The formula for the method of standard addition is:
Cs /(Cs + Ca)
= Ss / (Ss + Sa)
where:
Cs = concentration of the sample (unknown)
Ca = concentration of the addition (known)
Ss = instrument response from the sample (known)
Sa = instrument response from the addition (known)
Solving for Cs will give the correct concentration
for the analyte, corrected for multiplicative interferences. The
method of standard addition also has the extra benefit that it
will compensate for time drift of instrument response.
Another method of calibration that is useful in compensating
for temporal (i.e., time) drift of instrument response as well
as calibration curve nonlinearity is the method of bracketing.
It is basically a simplified direct calibration where only 2 standards
are used to establish the instrument response function. Bracketing
is used when the instrument response varies with time. In such
a case, a multi-point calibration line that was established at
the beginning of an analysis would be invalid during the analysis
because the slope of the calibration line changed while the samples
were being measured. Essentially, two calibration points are chosen
close to and on either side of the unknown sample signal and repeatedly
measured. The calculation is as follows:
CU = CL + (CH
– CL) x ((SU – SL)/(SH
– SL))
where:
CU = concentration of unknown
CL = concentration of low calibration standard
CH = concentration of high calibration standard
SU = instrument response from unknown
SL = instrument response from CL
SH = instrument response from CH
This may look complex, but all that is happening is that a
slope is established between the low and high standards and the
concentration of the unknown is determined based on the ratio
of (SU – SL)/(SH –
SL). Adding this concentration to the concentration
of the low standard will give the concentration of the unknown
sample.
To be continued with a discussion of internal standardization.